

Hypoplastisches Linksherzsyndrom (HLHS)
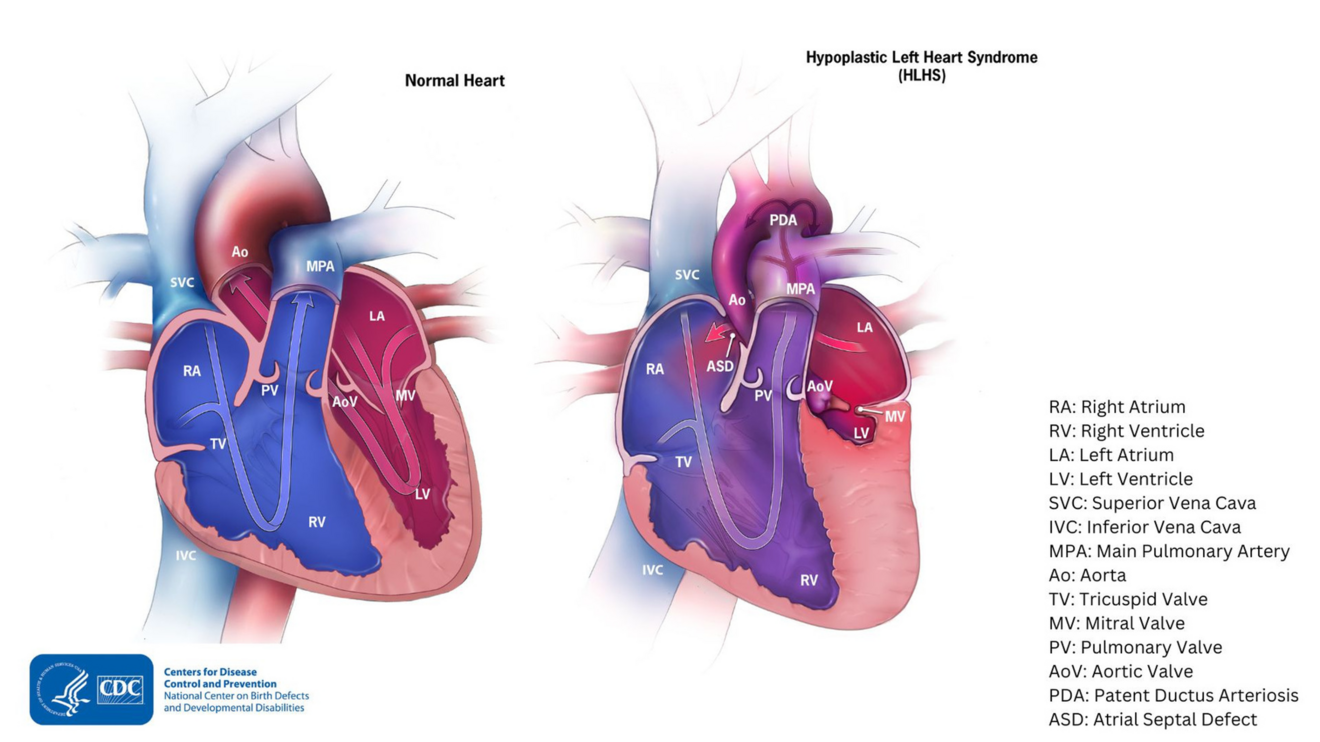
Beim Hypoplastischen Linksherzsyndrom (HLHS) sind die linken Herzkammern und die zugehörigen Strukturen stark unterentwickelt. Dadurch kann die linke Herzkammer den Körperkreislauf nicht oder nur unzureichend versorgen.
Typische Merkmale sind:
Starke Verengungen oder Verschlüsse an der Aortenklappe (Aortenstenose oder -atresie) und/oder Mitralklappe (Mitralstenose oder -atresie)
Eine stark verkümmerte linke Herzkammer (Hypoplasie oder Aplasie)
Eine Unterentwicklung (Hypoplasie) der aufsteigenden Aorta und des Aortenbogens bei intaktem Ventrikelseptum
Heutzutage wird der Begriff HLHS für besonders schwere Formen der Linkshypoplasie verwendet, bei denen der Körperkreislauf komplett von der rechten Herzkammer abhängig ist.
Symptome
Kinder mit HLHS zeigen häufig Blaufärbung der Haut (Zyanose), blasse Haut, schwache Durchblutung, schnelle Atmung und Herzschlag sowie schwache Pulse.
Ursache
Das sauerstoffreiche Blut kann nicht normal aus der linken Herzkammer in die Aorta fließen, da die Aortenklappe verengt oder verschlossen ist. Das Blut vermischt sich im rechten Herzen und wird dann sowohl zur Lunge als auch zum Körper geleitet. Nach der Geburt schließen sich normalerweise Verbindungen (Foramen ovale und Ductus arteriosus), was bei HLHS lebensbedrohlich ist, da der Körper dann kaum noch Blut erhält.
Die rechtzeitige Gabe von Prostaglandin hält den Ductus arteriosus offen und sichert so die Blutversorgung.
Prognose
Kurz- und mittelfristig ist die Prognose durch die komplexe Herzerkrankung mit nur einer funktionierenden Herzkammer (singulärer Ventrikel) oft gut. Die langfristige Entwicklung hängt stark von individuellen Faktoren und möglichen Komplikationen ab und ist schwer vorhersehbar.
Wichtige Informationen für Ärzt:innen
HLHS ist ein hochkomplexer, kritischer Herzfehler mit ductusabhängiger Systemzirkulation. Die Diagnose wird häufig schon pränatal im Rahmen einer Feindiagnostik gestellt. Eine umfassende Beratung der Eltern durch erfahrene Kinderkardiolog:innen ist entscheidend, um die Therapieoptionen zu besprechen.
Behandlung
Die lebensrettende Therapie umfasst eine Serie von Operationen. Die erste, die Norwood-Operation, erfolgt meist innerhalb der ersten Lebenswoche. Direkt nach der Geburt sollte das Neugeborene durch Spezialisten versorgt und die Diagnose mit Ultraschall bestätigt werden. Zur Stabilisierung wird Prostaglandin verabreicht, um den Ductus arteriosus offen zu halten. Anschließend erfolgt die Verlegung zur Operation.
Weitere Eingriffe erfolgen im Alter von 4–6 Monaten und rund mit 2 Jahren, um die sogenannte Fontan-Palliation zu erreichen.
Operationen im Überblick
Norwood-1-Operation
Umstrukturierung des Blutflusses, sodass die rechte Herzkammer den Körper- und Lungenkreislauf übernimmt. Die verengte Aorta wird erweitert und mit der Pulmonalarterie verbunden. Ein Shunt sorgt für die Lungenblutversorgung.
Glenn-Operation (Norwood-Stufe 2)
Im Alter von 3-5 Monaten wird die obere Hohlvene direkt mit der rechten Lungenschlagader verbunden, um die rechte Herzkammer zu entlasten.
Fontan-Operation (Norwood-Stufe 3)
Die untere Hohlvene wird ebenfalls an die Lungenschlagader angeschlossen, sodass Lungen- und Körperkreislauf komplett getrennt sind. Die rechte Herzkammer pumpt dann nur noch sauerstoffreiches Blut in den Körperkreislauf.